|
WISARD[wɪzərd] Workbench for Integrated Superfast Association study with Related Data |
|
Case tutorial
- Study design
- Data management
- Summary statistics
- Association analysis
- Relationship matrix
- Population stratification
- Association analysis
- Association analysis with population stratification
- Family-based association analysis
- Epistasis analysis
- Gene-level association
- Gene-level association with population stratification
- Family-based gene-level association
- Pathway-level association
- Meta-analysis
- Miscellaneous
Convert/split/merge
This section is about
- Conversion
- PLINK PED format
- Binary PED format
- Transposed PED format
- Long file format
- Number-coded format
- Variant Calling Format (VCF)
- GEN file format
- Binary GEN file format
- Conversion-related options
- Notation for missing phenotype
- Notation for missing genotype
- Notation for case/control phenotype
- Other exportable dataset
- Splitting input dataset
- Merging multiple files
Conversion [top]
WISARD can convert retrieved dataset into many other formats.
Below are supported file formats from WISARD and their description.
In below example,
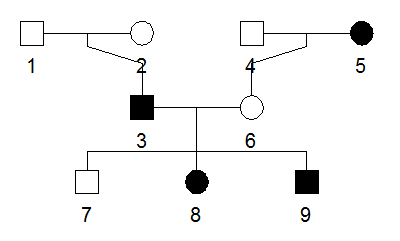
PLINK PED format
The option --makeped, generates .ped (genotype & pedigree, sex and single phenotype) and .map (marker info. without allele info.).
Binary PED format
The option --makebed, generates .bed (binary-coded genotype), .fam (pedigree, sex and phenotype), and .bim (marker info. with allele info.).
|
NOTE! |
In default, BED file is generated with SNP-major format. It can be transposed as individual-major format with --sampmajor option. |
Transposed PED format
The option --maketped, generates .tped (Marker info. and genotype) and .tfam (pedigree, sex and single phenotype).
Long file format
The option --makelgen, generates .lgen (FID, IID and genotype) and .map (marker info. without allele info.).
Number-coded format
Generates .raw (minor allele, number-coded genotype, pedigree, sex, and single phenotype) Available codings are additive (--makeraw), dominant (--makedom) and recessive (--makerec).
|
NOTE! |
In default, this format includes header. It can be omitted with --outnoheader option! |
|
NOTE! |
In default, this format exports six mandatory columns (FID, IID, parental IID, sex, and default phenotype). Phenotype can be separately exported with --outphenoonly option! |
Variant Calling Format (VCF)
The option --makevcf, generates .vcf (genotype and others) and .fam (pedigree, sex and phenotype). For details, see this page.
GEN file format
The option --makegen, generates .gen (allele info. and probability-coded genotype) and .sample (FID, IID and multiple phenotypes and covariates).
Binary GEN file format
With the option --makebgen, binary GEN format dataset is generated, made of .bgen (equivalent to .gen but binary-coded) and .sample (FID, IID and multiple phenotypes and covariates)
|
NOTE! |
In default, genotype probability data of Binary GEN file is not zipped, but stored in computationally efficient form. In order to reduce the size of dataset, --zipbgen option can be used to shrink it! |
Conversion-related options [top]
While performing conversion, details can be manipulated for the dataset with following options:
Notation for missing phenotype
Missing phenotype is exported as -9 in default. --outmispheno can alter this.
Notation for missing genotype
A default notation for missing genotype is 0, for most of files supported by WISARD, or '.' for VCF file. --outmisgeno can alter this.
|
NOTE! |
Some file formats with fixed notation for missing genotype is unaffected by this option! |
Notation for case/control phenotype
For dichotomous phenotype, they are exported as 2 (case/affected) and 1 (control/unaffected). --out1case and --outcact can alter this.
Other exportable dataset [top]
In addition to widely used file format, following subsets can be generated also:
- Phenotype file: First-line header for phenotype names and following phenotype value records equivalent to the number of samples in the dataset. (FID, IID and phenotypes)
- Covariates file: Same as phenotype file but covariates value records.
- Genotype file: A plain matrix file with n by p dimension, where n is number of samples and p is number of markers.
Splitting input dataset [top]
Sometimes, especially for the large-scale NGS dataset, the entire volume of dataset is too massive. In such situation, splitting the dataset with specific criterion could allow more flexible dataset handling. WISARD provides easy splitting functionality via --split option.
Above command produces chromosome-wise Binary PED file from test_miss0.bed.
Above command produces family-wise Binary PED file from test_miss0.bed.
Merging multiple files [top]
WISARD supports multiple file loading, via --merge option. Merging multiple files in WISARD assumes two components, (1) Base dataset, assigned by ordinary input-related options, and (2) Datasets to be merged with base dataset. --merge option assigns an information of second component, and let WISARD know that there is/are dataset(s) to be merged into. In order to use this function, an argument should be assigned to --merge option. It can be (1) sequence of paths divided by comma with no separator, or (2) a path of file containing multiple files to be merged. In case of (1), only one dataset can be assigned, while (2) can define multiple datasets. Every paths for --merge option must satisfy conditions below.
- Every file path should be made of absolute path or appropriate relative path
- Each dataset must be represented by a set of paths with given sequence ( Binary PED file format : bed, bim and fam )
- For the first file, its extension must be equivalent to requirement
- If following paths of dataset have exactly same name but extension, it can be omitted
- In case of (2), the contents of file must be set of lines, and each line must indicate single dataset
( PED file format : ped and map )
( Long file format : lgen and map )
( Transposed PED file format : tped and tfam )
( VCF format : vcf and fam )
|
NOTE! |
--merge does not export merged dataset itself like other options, unless any export-related option is assigned |
In order to allow more flexible merging options, --mergemode can be further utilized. Currently, below merging modes are possible. Note that this merging strategy only applies to the overlapped genotypes.
- Consensus mode (default): If the genotype in the base dataset is missing, it is replaced. If the non-missing genotype or replaced genotype is not concordant with following genotype, it is marked as NA and never replaced.
- Replace missing only: If the genotype in the dataset is missing, it is replaced. Otherwise do nothing.
- Replace all non-missing: If the genotype in the dataset to be merged, it is replaced. Otherwise do nothing.
- Do not replace at all: All data (any conflicting/missing/whatsoever overlap) is preserved.
- Replace without condition: Any overlapping genotype will be replaced.
Edit this page