HisCoM-PCA
BIBS lab, Seoul National University
Main Page Usage Download
Usage
HisCoM-PCA is written by R and can be installed by the following steps.
Step 1. Source Code Download
HisCoM-PCA can be run by R program. It also need WISARD program which can be download from To run the HisCoM-PCA, you should first download the Download zip file which contains ‘HisCoM-PCA.R’ and sample data.
Step 2. Input File for HisCoM-PCA
Four input files are required to run HisCoM-PCA. The following table shows the summary of four files.
| Genetic data file (.csv) | Contains SNP values coded as 0,1,2 (number of minor allele). |
| Gene-SNP annotation file (.set) | Contains two columns: (1) gene name and (2) SNP id. |
| Pathway-Gene annotation file (.set) | Contains two columns: (1) pathway name and (2) gene name. |
| Trait file (.txt) | Contains (1) phenotype and (2) covariates. |
Genetic file
Row represents the SNP ID and the column represents the sample id. Each cell represents the SNP value coded as 0,1,2 according to the number of minor allele in each locus. If the genetic data is in PLINK format, users can use PLINK to recode the SNP as 0, 1, 2.
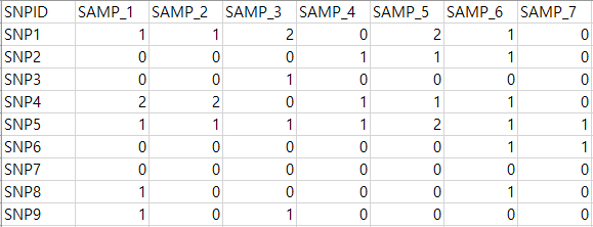
Gene-SNP annotation file
Each line consists of two columns for gene name and SNP id, respectively.
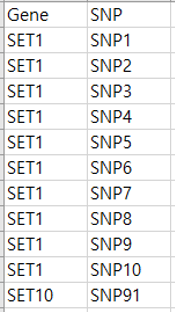
Pathway-Gene annotation file
Each line consists of two columns for pathway name and gene name, respectively. If the pathway-gene annotation file is download from a published database such as KEGG which is a row-wise format file, users can use the ‘Pathway_COLanno’ function in ‘HisCoM-PCA.R’ to transform it as the two-columns input format.
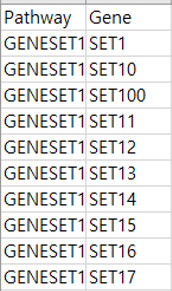
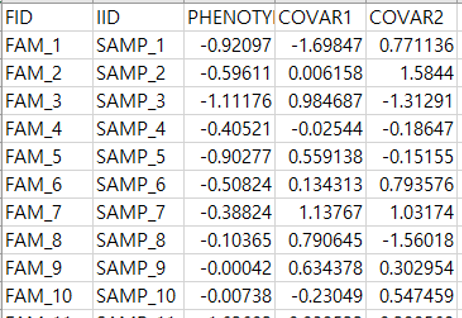
Trait file
First two columns must be family id (FID) and individual id (IID) and the column names for the first two columns are “FID” and “IID”. The IID must be same with the sample id in ‘Genetic file’. This file contains phenotype and all covariates which will be used in analysis. The column names are asked in this file.
Step 3. Running the sample data
Using four files from Step 2, HisCoM-PCA can be run. There are some parameters for main function of HisCoM-PCA.

Step 4. Results
The followings are the main output. More detail information about the other output files can be fined in http://statgen.snu.ac.kr/wisard/
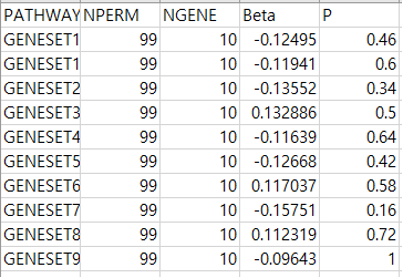
- Pathway results
- result.pathway.ressum.csv: Each line consists of 5 columns showing pathway name (PATHWAY), number of permutation (NPERM), number of genes in each pathway (NGENE), pathway coefficient (Beta), permutation p-value (P) respectively.
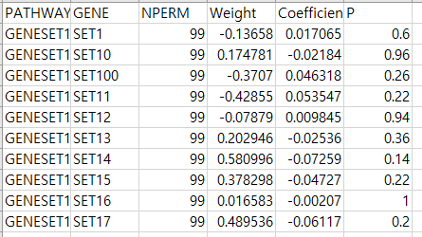
- Gene results
- result.gene.ressum.csv: Each line consists of 6 columns showing pathway name (PATHWAY), gene name (GENE), number of permutation (NPERM), weight of gene for the pathway (Weight), gene coefficient (Beta), permutation p-value (P) respectively.
The coefficient contained in this result file is about the weight (w_gene) value which represents the effect of the gene on the pathway.
Maintenance
If there is error, please let us know about the running problem to solve the problem and improve our program.
Contact : nn1123_94@126.com