HisCoM-PAGE
BIBS lab, Seoul National University
Main Page Usage Tutorial Download
Tutorial
To provide an information about running HisCoM-PAGE, we provide a short tutorial to run HisCoM-PAGE.
You can download the sample input file from the below link.
- To run the HisCoM-PAGE, you should first download theDownload zipfile which contains ‘program code.R’, data_preparation.R.
- In the tutorial step, you should download the ‘Tutorial’ zip file above which contains as follows: “toy_gene_expression.rds”, “toy_pathway.rds”, and “toy_trait” data file.
[1] Prepare the gene expression data and pathway database.
setwd(“/mydesktop/myfolder”)
exprs <-readRDS(“toy_gene_expression.rds”)
pathwayDB <-read.csv(“toy_pathway.csv”,header=T)
trait<-read.csv(“toy_trait.csv”,header=T)
[2] Using the pathway database and gene expression file which were loaded above step, make an input_gene expression file. Users need to make a gene expression file format which is described at the Step 3 of the manual of HisCoM-PAGE.
expression=c()
for (i in 1:ncol(pathwayDB)){
gene<- pathwayDB[,i]
geneup<-as.character(na.omit(gene))
probelist<-na.omit(match(geneup,rownames(exprs)))
pexp<-t(countdata2[probelist,]); expression=cbind(expression,pexp)
}
saveRDS(expression,"input_expression.rds")
Data parsing example code
[3] Using the pathway database, make an annotation file.
Users need to make a pathway annot file which is described at the Step 3 of the manual of HisCoM-PAGE. Through the following steps, you will ready to run HisCoM page function.
genelist=c()
pathname=c()
for(i in 1:ncol(list)){
gene=list[,i];geneup<-as.matrix(na.omit(gene))
genelist=rbind(genelist,geneup);count<-NROW(geneup)
l=colnames(list[i]);path=as.matrix(rep(c(l),count))
pathname=rbind(pathname,path)
}
symbolannot<-cbind(pathname,genelist)
dic<-data.frame(symbolannot)
colnames(dic)<-c("path","gene")
l<-unlist(unique(dic$path));m<-match(dic$path,as.character(l))
pathname<-paste("path",match(dic$path,as.character(l)),sep="")
symbolannot<-cbind(pathname,genelist)
genum=c()
for ( i in 1:length(l)){
N<-length(which(m==i));al<-paste("gene_",i,"_",1:N,sep="")
genum=c(genum,al)
}
dic$path<-pathname;dic$gene<-genum
saveRDS(dic,"annotation_input.rds")
[4] Run HisCoM-PAGE
The following is an example code to run the HisCoM-PAGE in R program.
setwd(“/mydesktop/myfolder”)
source("program code.R")
pathway_annot <-readRDS ("annotation_input.rds")
# load annotation file.
gene<-readRDS("input_expression.rds )
# load gene expression file.
y<-readRDS(“toy_trait”, header=T) # load survival time and status file.
result<- hiscom_page(gene,y,pathway_annot,iter_num=10000)
#iter_num : number of permtation.
The following screen will be shown in the console window after normal running. This step is optimal lambda searching step. Running time may vary, according to the number of genes and the number of samples.
After the previous step, this console window will be shown to your screen. It is a p-value calculation step.
Lastly, the final ‘result’ object will return 3 lists. List1 represents the pathway result, list 2 the gene results I, and list 3 the gene results II.
write.csv(result[1], “pathway_result.csv”)
write.csv(result[2], “gene1_result.csv”)
write.csv(result[3], “gene2_result.csv”)
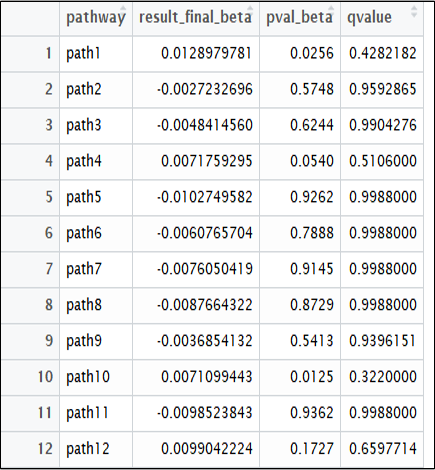
Fig 1. Pathway result
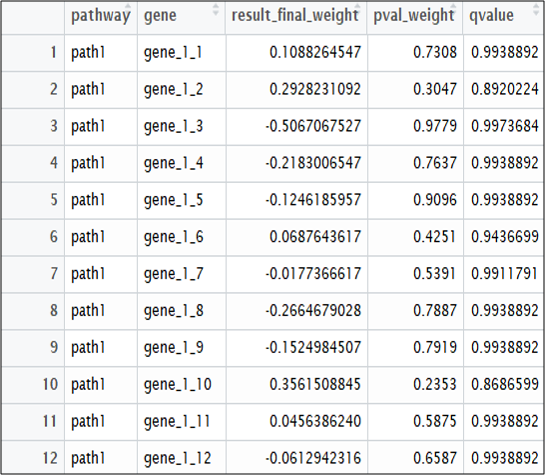
Fig 2. Gene-1 result
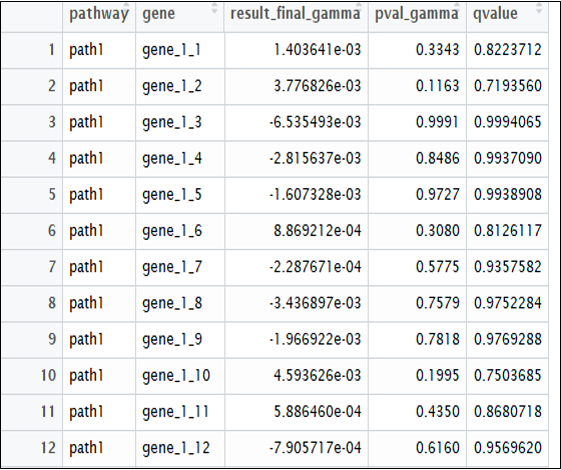
Fig 3. Gene-2 result
Maintenance
If there is error, please let us know about the running problem to solve the problem and improve our program.
Contact : lydiamok25@snu.ac.kr