HisCoM-Kernel
BIBS lab, Seoul National University
Main Page Usage (C/C++ version) Download
Usage (C/C++ version)
HisCoM-Kernel is written by R and can be installed by the following steps.
- Step 1. Source Code Download
- Step 2. Input Files for HisCoM-Kernel
- Step 3. Wisard command
- Step 4. Results
Step 1. Source Code Download
HisCoM-Kernel software written by C/C++ can be run by WISARD program. To run the HisCoM-Kernel, you should first download the program from http://statgen.snu.ac.kr/wisard/?act=download.
Step 2. Input Files for HisCoM-Kernel
As inputs, three data files are required: phenotype data file, omics data file, and pathway-biomarker (e.g. gene, metabolite) matching file. Example data with the three data files are included in the ‘ExampleC_Input’zip file.
|
Pathway-biomarker matching file (.set) |
Contains (1) pathway name and (2) biomarker name. It should be made in the same format as ‘ExampleC_pathway_matching.set’ file. |
|
Omics data file (.raw) |
Contains gene expression or metabolite level values. The format is same as the ‘ExampleC_data.raw’file. |
|
Phenotype data file (.txt) |
Contains (1) sample ID and (2) Phenotype. To add covariates to the model, this file should contain covariate information. The format is same as the ‘ExampleC_ID_pheno.txt’file. |
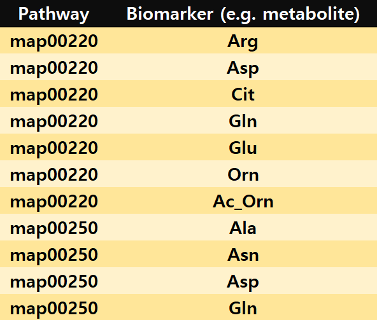
Pathway-biomarker matching file
Each line consists of two columns for pathway name and biomarker name, respectively. Here, biomarkers can be genes or metabolites. When reading the matching file, there should be no column names such as “ExampleC_pathway_matching.set” file.
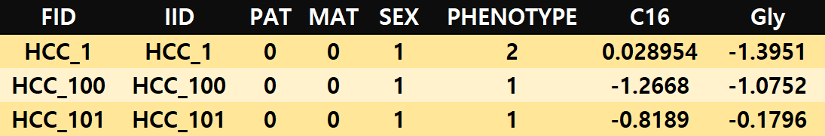
Omics data file
The first six columns have a format consistent with PLINK’s covariate table. Specifically, FID, IID, PAT, MAT, SEX, and PHENOTYPE indicate family ID, individual ID, paternal individual ID, maternal individual ID, sex (1=male, 2=female), and binary outcome (control=1, case=2). Starting from the 7th column, biomarker (e.g. gene, metabolite) name should be included. Each cell represents the gene expression or metabolite level value for each individual ID.
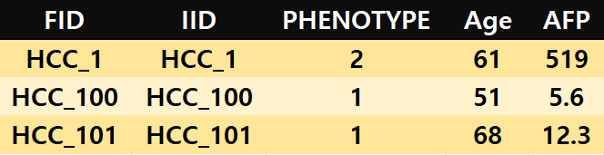
Trait file
Each row consists of two columns for family ID, individual ID, phenotypic information, and covariates, respectively. The family ID and individual ID should match the ID column order in the omics data file. For phenotype (i.e. binary outcome), case = 2, control = 1. Covariates should be included starting with the 3rd column. The example below shows age and AFP as covariates.
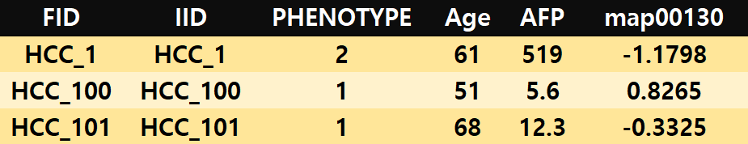
If you have any pathways that contain only a single biomarker, these pathways should be included after covariates, as in the example below.
Step 3. Wisard command
wisard --expression ExampleC_data.raw --pharaoh --hiscomkernel --sampvar ExampleC_ID_pheno.txt --pname PHENOTYPE --geneset ExampleC_pathway_matching --nperm 1000 --prolambda 500 –verbose –out test
- --hiscomkernel: By default, it uses Gaussian kernel. To use polynomial or linear kernel, you can use “--hiscomkernel polynomial” and “--hiscomkernel linear”.
- --pname: Column name indicating the phenotype
- --nperm: The number of permutation tests to calculate p-value
- --prolambda: Optimal lambda used to estimate beta coefficients of HisCoM-Kernel
- --out: Output file name can be specified.
- --hiscomkernelrho: For Gaussian kernel, the parameter ρ can be determined by adding an option as in the example “--hiscomkernelrho 1” (by default, ρ=2).
Step 4. Results
Following the tutorial in Step 3, the outputs are as follows:
- res.pharaoh.trueA: It provides beta coefficients representing pathway effects.
- res.pharaoh.permA: It provides a beta coefficient file for each permutation
- res.pharaoh.pathway.res: It provides p-values based on permutation tests for all pathways. The P_PHARAOH_SC column includes the p-value for each pathway.
Maintenance
If there is error, please let us know about the running problem to solve the problem and improve our program.
Contact : suhyun8695@gmail.com