Step 1. Execution
Step 2. Analyze dataset
Step 3. Advanced running
Step 1. Execution
Basically, PHARAOH-multi is a command-line software; i.e., it is NOT possible to run PHARAOH visually or interactive, like graphical user interface.
To provide an information about running PHARAOH, here we describe a short tutorial to run PHARAOH in your desktop or server.
Please proceed to the tutorial by the operating system that PHARAOH is running on.
Microsoft Windows
PHARAOH-multi supports from Windows XP (which is very legacy version of Windows, hence we really not recommend to run PHARAOH-multi on Windows XP, although we supports this version) to Windows 10.
In this tutorial, we describe how run the PHARAOH-multi software and do an analysis using PHARAOH-multi.
Step 1: Run command-line interface
The PHARAOH-multi software is command-line software. Hence it is required to run command-line interface to run the PHARAOH-multi software.
Run the command-line interface of Microsoft Windows by the following procedures.
- Type Windows key (
or
or
, depending on your keyboard appearance) + R.
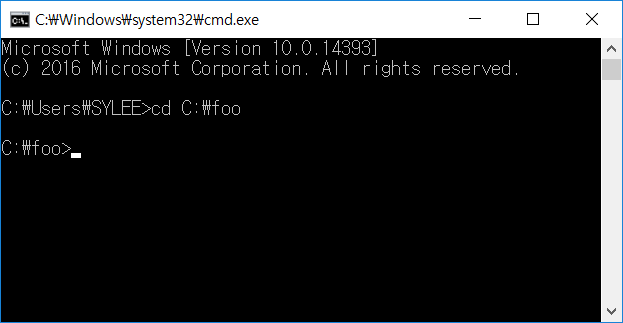
- Type cmd to launch the command-line prompt.After executing the command-line, move to the directory that the PHARAOH-multi program was extracted. Let’s say the directory is C:\foo.
Figure 1. Type cd C:\foo to move the directory.
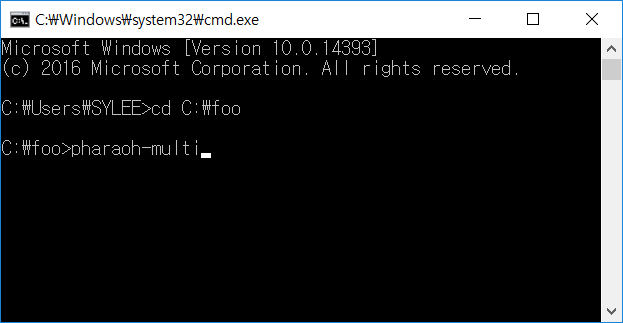
If there is the PHARAOH-multi program was extracted properly, type the following command will showing proper output and generating one file, res.log.
Figure 2. Type pharaoh-multi to execute the PHARAOH program.
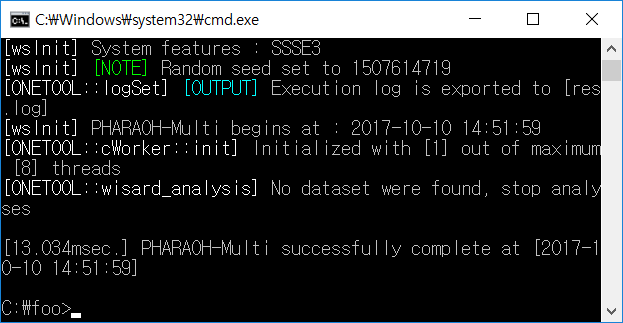
Figure 3. When the PHARAOH-multi program has executed properly.
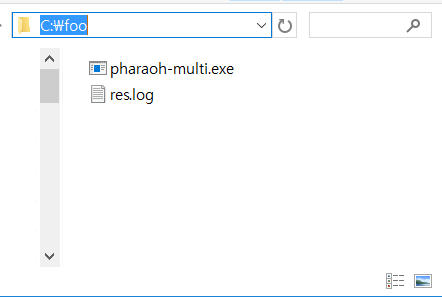
If the above Figure 3 does not shown, it indicates that there was a problem before the preparation of the PHARAOH-multi program. However, unless there was no problem, res.log file will be shown in C:/foo directory.
Figure 4. res.log has created.
The generated log file contains summary of the performed analysis, and helpful information to find out and solve the problem.
Now, the preparation has over, let’s do some analysis using PHARAOH-multi.
Linux distributions
Note 1: In this manual, the command-line interface is assumed.
Note 2: An appearance of command-line interface may differ with your one.Let’s start from the initial screen of terminal, and go to the directory where the PHARAOH program was extracted.
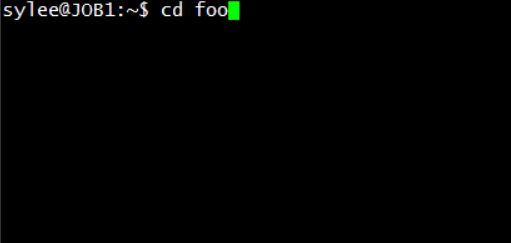
Figure 1. Go to the directory of the PHARAOH program
Check whether the PHARAOH program is in the path or not.
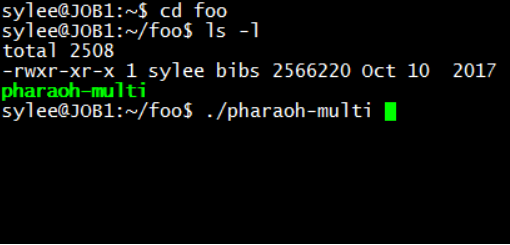
Figure 2. Check and run the PHARAOH program
As shown in Figure 2, there is a file named PHARAOH, and its permission (leftmost part of 4th line) is correct. If the permission is not correct, the program will not be executed.
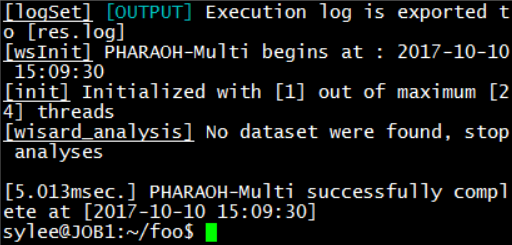
Type ./pharaoh to execute the program. Note that the program will not be executed if the first two characters(dot and slash) are omitted.Figure 3. Successful execution
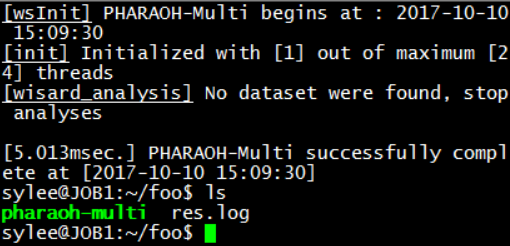
Figure 4. Check the log file
Step 2. Analyze dataset
[expand title="Click to see the contents"]
We provide a sample dataset to try the PHARAOH-multi program.
The sample dataset can be downloaded from the below links.
pharaoh_multi sample dataset (tar.gz format)
pharaoh_multi sample dataset (zip format)
The sample dataset consists of five files: pharaoh_multi.bed, pharaoh_multi.fam, pharaoh_multi.bim, pharaoh_multi_pheno.txt, pharaoh_multi_gene.txt and pharaoh_multi_pathway.txt.
| pharaoh_multi.bed | Contains genotypes of the samples (binary coded). |
| pharaoh_multi.bim | Contains information of genetic variants. |
| pharaoh_multi.fam | Contains information of pedigree. |
| pharaoh_multi_gene.txt | Contains the mapping between the genetic variants and the gene. |
| pharaoh_multi_pathway.set | Contains the mapping between the genes and the pathway. |
| pharaoh_multi_pheno.txt | Contains the phenotypes of the samples. |
Let’s do the simplest run using the PHARAOH-multi program.
Preparation
After downloading the sample dataset, it is required to uncompress the downloaded files to the directory where the PHARAOH-multi program was extracted.
Note: For Microsoft Windows, we strongly recommend to use other GUI-based archive software, such as BandiZip.
The following commands for Linux distributions will extract the sample dataset into the directory where the zipped file exists.
# For Linux distributions (zip) pharaoh@JOB1:~/foo$ unzip pharaoh_multi_dataset.zip # For Linux distributions (tar.gz) pharaoh@JOB1:~/foo$ tar xvfz pharaoh_multi_dataset.tar.gz
Full-automatic running
In full-automatic mode, only two parameters that PHARAOH-multi requires are
- (–cv) Number of cross-validations to perform to determine an optimal lambda(λ) value.
- (–pname) Phenotypes that are included to the analysis.
- (–nperm) Number of permutations to get the p-values. In default, 1000 is recommended value.
In this mode, the PHARAOH-multi program first determines an optimal value of λ via cross-validation, and next run PHARAOH-multi analysis.
# For Linux distributions pharaoh@JOB1:~/foo$ ./pharaoh-multi --pharaoh --cv 2 --nperm 1000 --bed pharaoh_multi.bed --set pharaoh_multi_gene.txt --geneset pharaoh_multi_pathway.txt --pheno pharaoh_multi_pheno.txt --pname PHENOTYPE1,PHENOTYPE2 --out test_res # For Microsoft Windows C:\foo> pharaoh-multi --pharaoh --cv 2 --nperm 1000 --bed pharaoh_multi.bed --set pharaoh_multi_gene.txt --geneset pharaoh_multi_pathway.txt --pheno pharaoh_multi_pheno.txt --pname PHENOTYPE1,PHENOTYPE2 --out test_res–pharaoh command let the PHARAOH-multi program performs PHARAOH-multi analysis.
–cv and –nperm are the parameter that are required to perform PHARAOH-multi analysis.
–bed, –sampvar, –pname, –set and –geneset indicate the dataset, variants-gene mapping and genes-pathway mapping, respectively.
–out determines the prefix of results.
Results
The PHARAOH-multi program generates the following files after the normal run.
[prefix].pharaoh.multi.res The main result file. The details of the output files are as follows.
Note that the columns with red color should be focused on the interpretation.
[prefix].pharaoh.pathway.res Column name Description PATHWAY Name of pathway NPERM Number of performed permutations NGENE Number of genes included in the pathway NVARIANT Total number of collapsed rare variants in the pathway P_KOST Combined-type multivariate p-value of the pathway P_MULTI Joint-type multivariate p-value of the pathway
[/expand]
Step 3. Advanced running
[expand title="Click to see the contents"]
Fix ridge penalty value λ
If there is a known value of ridge penalty or is required to fine-grained tuning of the result, a fixed penalty value λ can be used.
- (–prolambda) Pre-determined lambda(λ) value. –cv option will not required if use this option.
The following example demonstrates usage of fixed penalty value λ.
# For Linux distributions pharaoh@JOB1:~/foo$ ./pharaoh-multi --pharaoh --nperm 1000 --bed pharaoh_multi.bed --set pharaoh_multi_gene.txt --geneset pharaoh_multi_pathway.txt --pheno pharaoh_multi_pheno.txt --pname PHENOTYPE1,PHENOTYPE2 --prolambda 155 # For Microsoft Windows C:\foo> pharaoh --pharaoh --nperm 1000 --bed pharaoh_multi.bed --set pharaoh_multi_gene.txt --geneset pharaoh_multi_pathway.txt --pheno pharaoh_multi_pheno.txt --pname PHENOTYPE1,PHENOTYPE2 --prolambda 155
–pharaoh command let the PHARAOH-multi program performs PHARAOH-multi analysis.
–prolambda fixes the λ parameter (penalty parameter).
–ped, –sampvar, –pname, –set and –geneset indicate the dataset, variants-gene mapping and genes-pathway mapping, respectively.
Apply user-defined weight on genetic variants
Recent advancement in bioinformatics allows multiple strategy on variant collapsing with various bioinformatics resources, such as predicted effect on protein structure. For this case, users can allow user-defined weight on genetic variants using PHARAOH-multi, with the below command.
# For Linux distributions pharaoh@JOB1:~/foo$ ./pharaoh --weight weight.txt --pharaoh --nperm 1000 --bed pharaoh_multi.bed --set pharaoh_multi_gene.txt --geneset pharaoh_multi_pathway.txt --pheno pharaoh_multi_pheno.txt --pname PHENOTYPE1,PHENOTYPE2 # For Microsoft Windows C:\foo> pharaoh --weight weight.txt --pharaoh --nperm 1000 --bed pharaoh_multi.bed --set pharaoh_multi_gene.txt --geneset pharaoh_multi_pathway.txt --pheno pharaoh_multi_pheno.txt --pname PHENOTYPE1,PHENOTYPE2
–pharaoh command let the PHARAOH-multi program performs PHARAOH-multi analysis.
–weight assigns user-defined weight on the variants, as a file consists of two columns.
In the file, the first column indicates the variant, and the second column contains “user-defined weight” when collapsing variants into the gene.
–ped, –sampvar, –pname, –set and –geneset indicate the dataset, variants-gene mapping and genes-pathway mapping, respectively.
[/expand]